Background
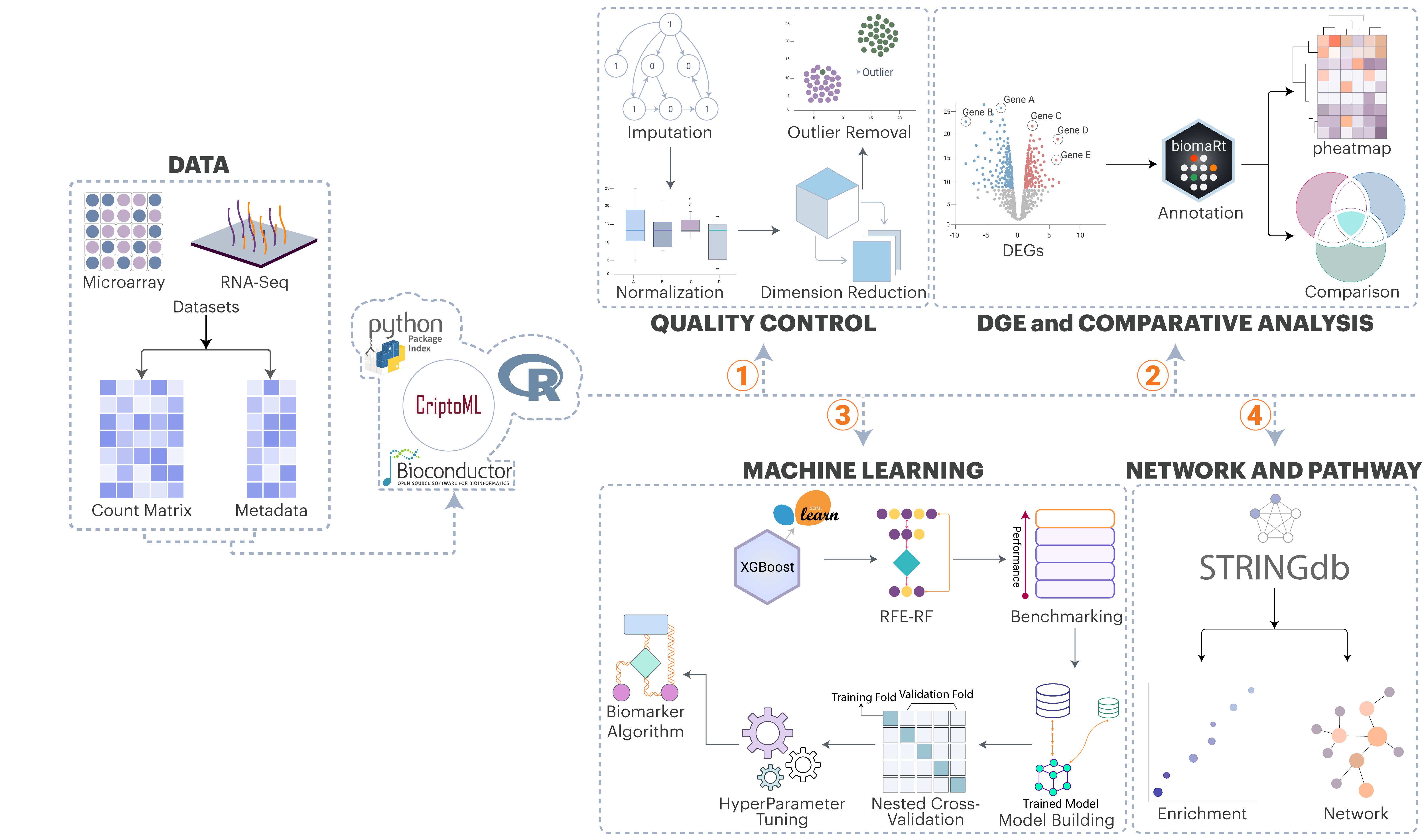
Transcriptomic biomarker discovery has been a challenge due to variation in datasets and platforms, complexity in statistical and computational methods, integration of multiple programing languages, intricacy of ML workflow to evaluate biomarkers. Standard workflows necessitate several stages (quality control, normalization, differential expression), typically executed in R or Python, resulting in bottlenecks for non-experts.
Method
We present omicML, an intuitive graphical user interface (GUI) that combines transcriptomic data analysis with machine learning (ML)-based classification via integrating R and Python packages/libraries. It supports both RNA-Seq and microarray data, automating preprocessing and differential expression analysis. Our extensive ML pipeline enables both supervised and unsupervised learning, integrates various datasets based on candidate gene signatures, and systematically finalizes the biomarker algorithm.
Result
In a case study, omicML identified a six-gene diagnostic model that distinguishes Mpox (monkeypox virus) infections from those caused by other viruses, including SARS-CoV-2, HIV, Ebola, and varicella-zoster. These results illustrate omicML's capacity to discern clinically relevant biomarkers from complex transcriptome data.
Conclusion
Integrating data normalization, differential gene expression analysis, annotation, heatmap analysis, dataset integration, batch effect removal, machine learning analysis, and functional analysis into a unified system diminishes technical barriers and accelerates the conversion of expression data into diagnostic insights for clinicians and bench scientists.